if (!requireNamespace("fgsea", quietly = TRUE)) {
BiocManager::install("fgsea")
}
if (!requireNamespace("msigdbr", quietly = TRUE)) {
install.packages("msigdbr")
}
library(bixverse)
library(data.table)
library(magrittr)Gene Set Enrichment Methods
Gene Set Enrichment Methods in bixverse
This vignette shows you how to use the gene set enrichment methods implemented in bixverse.
Intro
Gene set enrichment analysis sits at the heart of most functional genomics workflows. Given a set of genes of interest (or a ranked list of all measured genes) you want to know which biological processes, pathways, or molecular functions are over-represented relative to what you would expect by chance. bixverse provides Rust-accelerated implementations of the two dominant paradigms for this, along with gene ontology-aware variants of each.
Over-representation analysis (ORA) via the hypergeometric test asks: given that I have drawn genes from a universe of , and of those belong to a particular gene set, is the overlap of larger than expected? This is fast and interpretable, but requires a discrete gene list — typically genes passing some fold-change and significance threshold. The choice of threshold and universe can substantially affect results, which is a known limitation of the approach.
Gene Set Enrichment Analysis (GSEA) sidesteps this by working with a continuously ranked list of all measured genes, asking whether the members of a gene set are systematically concentrated towards the top or bottom of the ranking. This avoids arbitrary thresholding and tends to be more sensitive to coordinated but modest shifts in expression, at the cost of being more computationally demanding and less straightforward to interpret.
A practical complication with both approaches is the redundancy of the Gene Ontology (GO). The GO is a directed acyclic graph: a significant hit on a specific term (e.g., “mitochondrial complex I assembly”) will typically also produce significant hits on all of its ancestor terms (e.g., “oxidative phosphorylation”, “metabolic process”), most of which add no new information. bixverse provides two strategies for dealing with this: the elimination method of Adrian et al., which propagates signal up the DAG while removing explained genes, and a post-hoc simplification approach based on Wang semantic similarity, which collapses redundant terms after running a standard enrichment.
All methods are implemented in Rust and exposed via extendr. The performance gains relative to existing R implementations range from modest (~20% faster for GSEA relative to fgsea’s Rcpp implementation) to substantial: the parallelised hypergeometric test over large gene set libraries can be orders of magnitude faster than a naive R loop.
Hypergeometric tests
bixverse implements the standard hypergeometric over-representation test. In this example we load the Hallmark gene sets from msigdbr and construct a named list, which is the expected input format throughout bixverse for gene set analyses.
To test a single target gene set, use gse_hypergeometric. The gene universe defaults to the union of all genes represented in the gene_set_list if not provided explicitly. This is usually reasonable but worth checking if your assay covers a non-standard set of genes. FDR threshold and minimum overlap are also configurable.
results <- gse_hypergeometric(
target_genes = target_genes_1,
gene_set_list = h_gene_sets_ls
)
head(results)
#> gene_set_name odds_ratios pvals fdr hits
#> <char> <num> <num> <num> <num>
#> 1: HALLMARK_MYC_TARGETS_V2 170.67009 5.919144e-41 2.959572e-39 27
#> 2: HALLMARK_MYC_TARGETS_V1 35.36813 2.110201e-27 5.275502e-26 29
#> 3: HALLMARK_E2F_TARGETS 4.21748 1.448776e-03 2.414627e-02 8
#> gene_set_lengths target_set_lengths
#> <num> <int>
#> 1: 58 50
#> 2: 200 50
#> 3: 200 50When you have many target gene sets to test simultaneously, use gse_hypergeometric_list. Under the hood this runs all tests in parallel via rayon and optimiser FxHashSets, making it very fast even at scale.
target_genes_2 <- c(
sample(h_gene_sets_ls[["HALLMARK_TNFA_SIGNALING_VIA_NFKB"]], 20),
sample(h_gene_sets_ls[["HALLMARK_IL6_JAK_STAT3_SIGNALING"]], 25)
)
target_list <- list(
set_1 = target_genes_1,
set_2 = target_genes_2,
set_3 = c("random gene 1", "random gene 2", "random gene 3")
)
results_multiple <- gse_hypergeometric_list(
target_genes_list = target_list,
gene_set_list = h_gene_sets_ls
)
head(results_multiple)
#> target_set_name odds_ratios pvals fdr hits gene_set_lengths
#> <char> <num> <num> <num> <num> <num>
#> 1: set_2 165.038462 3.137168e-43 1.568584e-41 30 90
#> 2: set_1 170.788856 5.811282e-41 2.905641e-39 27 58
#> 3: set_1 35.393567 2.069809e-27 5.174524e-26 29 200
#> 4: set_2 29.964115 5.979167e-22 1.494792e-20 24 200
#> 5: set_2 11.995968 2.338661e-10 3.897768e-09 15 201
#> 6: set_2 8.571723 2.515351e-07 3.144189e-06 12 200
#> gene_set_name target_set_lengths
#> <char> <int>
#> 1: HALLMARK_IL6_JAK_STAT3_SIGNALING 45
#> 2: HALLMARK_MYC_TARGETS_V2 50
#> 3: HALLMARK_MYC_TARGETS_V1 50
#> 4: HALLMARK_TNFA_SIGNALING_VIA_NFKB 45
#> 5: HALLMARK_INFLAMMATORY_RESPONSE 45
#> 6: HALLMARK_INTERFERON_GAMMA_RESPONSE 45To give a sense of the scale this handles comfortably, the following runs 250 target gene sets against 5,000 gene sets drawn from a universe of 20,000 genes — 1.25 million hypergeometric tests in total. On most systems this completes in a few seconds.
seed = 10101L
set.seed(seed)
universe <- sprintf("gene_%i", 1:20000)
gene_sets_no <- 5000
target_gene_sets_no <- 250
gene_sets <- purrr::map(
1:gene_sets_no,
~ {
set.seed(seed + .x + 1)
size <- sample(20:100, 1)
sample(universe, size, replace = FALSE)
}
)
names(gene_sets) <- purrr::map_chr(
1:gene_sets_no,
~ {
set.seed(seed + .x + 1)
paste(sample(LETTERS, 3), collapse = "")
}
)
target_gene_sets <- purrr::map(
1:target_gene_sets_no,
~ {
set.seed(.x * seed)
size <- sample(50:100, 1)
sample(universe, size, replace = FALSE)
}
)
names(target_gene_sets) <- purrr::map_chr(
1:target_gene_sets_no,
~ {
set.seed(seed + .x + 1)
paste(sample(letters, 3), collapse = "")
}
)
tictoc::tic()
rs_results_example <- gse_hypergeometric_list(
target_genes_list = target_gene_sets,
gene_set_list = gene_sets
)
tictoc::toc()
#> 0.483 sec elapsedGene Ontology-aware enrichment: the elimination method
The naive approach of running a hypergeometric test independently for each GO term produces heavily redundant results, because significant child terms mechanically inflate their ancestors. The elimination method from Adrian et al. addresses this by traversing the ontology from the most specific terms (leaves) upwards. When a term reaches the significance threshold, the genes annotated to that term are removed from all of its ancestor terms before their tests are computed. This means the ancestor terms are tested on the residual signal not already explained by their more specific descendants, yielding results that are both less redundant and more interpretable.
bixverse ships with the human GO data and exposes it via a dedicated S7 class that transfers the ontology structure into Rust for efficient traversal.
go_data_dt <- get_go_data_human()
#> Loading the data from the package.
#> Processing data for the gene_ontology class.
#> Warning: The `father` argument of `dfs()` is deprecated as of igraph 2.2.0.
#> ℹ Please use the `parent` argument instead.
#> ℹ The deprecated feature was likely used in the bixverse package.
#> Please report the issue to the authors.
go_data_s7 <- GeneOntologyElim(go_data_dt, min_genes = 3L)The interface mirrors the generic hypergeometric functions: a single target set or a list, with Rust’s ownership model ensuring the ontology structure is shared across all tests without copying, keeping memory overhead low.
go_aware_res <- gse_go_elim_method(
object = go_data_s7,
target_genes = target_genes_1
)
head(go_aware_res)
#> go_id go_name odds_ratios
#> <char> <char> <num>
#> 1: GO:0003723 RNA binding 14.812443
#> 2: GO:0032040 small-subunit processome 44.202960
#> 3: GO:0042274 ribosomal small subunit biogenesis 43.541132
#> 4: GO:0005730 nucleolus 6.009426
#> 5: GO:0000055 ribosomal large subunit export from nucleus 455.891304
#> 6: GO:0031428 box C/D methylation guide snoRNP complex 455.891304
#> pvals fdr hits gene_set_lengths
#> <num> <num> <num> <num>
#> 1: 3.122680e-16 3.441817e-12 23 1205
#> 2: 1.623943e-08 6.489265e-05 6 72
#> 3: 1.766267e-08 6.489265e-05 6 73
#> 4: 8.523856e-08 2.348748e-04 18 1866
#> 5: 2.368116e-07 4.350230e-04 3 6
#> 6: 2.368116e-07 4.350230e-04 3 6And the version for lists:
go_aware_res_2 <- gse_go_elim_method_list(
object = go_data_s7,
target_gene_list = target_list
)
head(go_aware_res_2)
#> target_set_name go_name
#> <char> <char>
#> 1: set_1 RNA binding
#> 2: set_2 cell surface receptor signaling pathway via JAK-STAT
#> 3: set_2 inflammatory response
#> 4: set_2 cytokine activity
#> 5: set_2 chemokine activity
#> 6: set_2 external side of plasma membrane
#> go_id odds_ratios pvals fdr hits gene_set_lengths
#> <char> <num> <num> <num> <num> <num>
#> 1: GO:0003723 14.81244 3.122680e-16 3.441817e-12 23 1205
#> 2: GO:0007259 97.16563 8.704402e-15 9.588769e-11 9 66
#> 3: GO:0006954 22.90818 1.408854e-13 7.759966e-10 14 447
#> 4: GO:0005125 31.85212 1.130962e-12 4.152892e-09 11 235
#> 5: GO:0008009 99.30420 6.201736e-12 1.707958e-08 7 48
#> 6: GO:0009897 21.74176 9.192276e-12 2.025242e-08 12 379The DAG traversal is inherently sequential per gene set (each level depends on the one below), but tests within a level are parallelised via rayon. Running 100 target gene sets against the full human GO completes in a matter of seconds:
go_gene_universe <- unique(unlist(go_data_dt$ensembl_id))
go_target_sets_no <- 100L
seed <- 246L
go_target_gene_sets <- purrr::map(
1:go_target_sets_no,
~ {
set.seed(.x * seed)
size <- sample(50:100, 1)
sample(go_gene_universe, size, replace = FALSE)
}
)
names(go_target_gene_sets) <- purrr::map_chr(
1:go_target_sets_no,
~ {
set.seed(seed + .x + 1)
paste(sample(letters, 3), collapse = "")
}
)
tictoc::tic()
rs_results_example <- gse_go_elim_method_list(
object = go_data_s7,
target_gene_list = go_target_gene_sets
)
tictoc::toc()
#> 1.374 sec elapsedAlternative: post-hoc simplification of GO results
An alternative strategy is to run an unconstrained hypergeometric test over all GO terms and then collapse redundant results afterwards using semantic similarity. This approach is more flexible — you can apply it to any enrichment results, not just those produced by bixverse — but it does not adjust the test statistics themselves the way the elimination method does. Whether you prefer pre-hoc elimination or post-hoc simplification depends on your use case; for exploratory analyses the simplification approach can be more convenient, while for rigorous GO reporting the elimination method is generally preferable.
go_data <- load_go_human_data()
min_genes <- 3L
go_genes <- go_data$go_to_genes
go_genes_ls <- split(go_genes$ensembl_id, go_genes$go_id)
go_genes_ls <- purrr::keep(go_genes_ls, \(x) length(x) > min_genes)
go_results_unfiltered <- gse_hypergeometric(
target_genes = target_genes_1,
gene_set_list = go_genes_ls
)
head(go_results_unfiltered)
#> gene_set_name odds_ratios pvals fdr hits gene_set_lengths
#> <char> <num> <num> <num> <num> <num>
#> 1: GO:0003723 22.120609 1.723343e-23 1.579099e-19 31 1546
#> 2: GO:0006364 35.837205 2.936032e-12 1.345143e-08 10 159
#> 3: GO:0005730 8.860092 6.601460e-12 1.581347e-08 23 1927
#> 4: GO:0042254 43.069954 6.903186e-12 1.581347e-08 9 118
#> 5: GO:0005634 8.314215 9.730968e-12 1.783297e-08 39 6736
#> 6: GO:0005654 6.752334 9.279447e-11 1.417126e-07 30 4005
#> target_set_lengths
#> <int>
#> 1: 50
#> 2: 50
#> 3: 50
#> 4: 50
#> 5: 50
#> 6: 50The simplification uses Wang semantic similarity to group related terms, then retains the term with the best test statistic within each cluster. Ties on FDR are broken by ontology depth, preferring the more specific term.
go_parent_child_dt <- go_data$gene_ontology[
relationship %in% c("is_a", "part_of")
] %>%
setnames(
old = c("from", "to", "relationship"),
new = c("parent", "child", "type")
)
go_results_simplified <- simplify_hypergeom_res(
res = go_results_unfiltered,
parent_child_dt = go_parent_child_dt,
weights = setNames(c(0.8, 0.6), c("is_a", "part_of"))
)
head(go_results_simplified)
#> gene_set_name odds_ratios pvals fdr hits gene_set_lengths
#> <char> <num> <num> <num> <num> <num>
#> 1: GO:0003723 22.12061 1.723343e-23 1.579099e-19 31 1546
#> target_set_lengths
#> <int>
#> 1: 50GSEA
bixverse implements GSEA following Subramanian et al. for testing against a continuously ranked gene list. The package also wraps the fgsea multilevel method from Korotkevich et al., which uses an adaptive permutation scheme to estimate precise p-values efficiently. Both are available for comparison.
library("fgsea")
data(examplePathways)
data(exampleRanks)
set.seed(42L)
fgsea_res <- fgsea(
pathways = examplePathways,
stats = exampleRanks,
minSize = 15,
maxSize = 500
) %>%
setorder(pathway)
head(fgsea_res)
#> pathway
#> <char>
#> 1: 1221633_Meiotic_Synapsis
#> 2: 1445146_Translocation_of_Glut4_to_the_Plasma_Membrane
#> 3: 442533_Transcriptional_Regulation_of_Adipocyte_Differentiation_in_3T3-L1_Pre-adipocytes
#> 4: 508751_Circadian_Clock
#> 5: 5334727_Mus_musculus_biological_processes
#> 6: 573389_NoRC_negatively_regulates_rRNA_expression
#> pval padj log2err ES NES size
#> <num> <num> <num> <num> <num> <int>
#> 1: 0.5490534 0.7262873 0.06674261 0.2885754 0.9399884 27
#> 2: 0.6952862 0.8366277 0.05445560 0.2387284 0.8366856 39
#> 3: 0.1122449 0.2599823 0.21392786 -0.3640706 -1.3460572 31
#> 4: 0.7826888 0.8799951 0.05312981 0.2516324 0.7287088 17
#> 5: 0.3580060 0.5579562 0.08197788 0.2469065 1.0498921 106
#> 6: 0.4198895 0.6197865 0.08407456 0.3607407 1.0446784 17
#> leadingEdge
#> <list>
#> 1: 15270,12189,71846
#> 2: 17918,19341,20336,22628,22627,20619,...[10]
#> 3: 76199,19014,26896,229003,17977,17978,...[12]
#> 4: 20893,59027
#> 5: 60406,19361,15270,20893,12189,68240,...[12]
#> 6: 60406,20018How does bixverse look in comparison?
bixverse_fgsea <- calc_fgsea(
stats = exampleRanks,
pathways = examplePathways,
gsea_params = params_gsea(min_size = 15L)
) %>%
setorder(pathway_name)
head(bixverse_fgsea)
#> pathway_name
#> <char>
#> 1: 1221633_Meiotic_Synapsis
#> 2: 1445146_Translocation_of_Glut4_to_the_Plasma_Membrane
#> 3: 442533_Transcriptional_Regulation_of_Adipocyte_Differentiation_in_3T3-L1_Pre-adipocytes
#> 4: 508751_Circadian_Clock
#> 5: 5334727_Mus_musculus_biological_processes
#> 6: 573389_NoRC_negatively_regulates_rRNA_expression
#> es nes pvals fdr
#> <num> <num> <num> <num>
#> 1: 0.2885754 0.9321129 0.5588723 0.7359532
#> 2: 0.2387284 0.8353752 0.7140600 0.8557113
#> 3: -0.3640706 -1.3241710 0.1282051 0.2900703
#> 4: 0.2516324 0.7211793 0.8109541 0.9063587
#> 5: 0.2469065 1.0551934 0.3751804 0.5816288
#> 6: 0.3607407 1.0338840 0.4204947 0.6207015
#> leading_edge n_more_extreme log2err
#> <list> <num> <num>
#> 1: 15270,12189,71846,19357 336 0.06407038
#> 2: 17918,19341,20336,22628,22627,20619,...[15] 451 0.05029481
#> 3: 76199,19014,26896,229003,17977,17978,...[12] 49 0.19991523
#> 4: 20893,59027,19883 458 0.04959020
#> 5: 60406,19361,15270,20893,12189,68240,...[13] 259 0.07707367
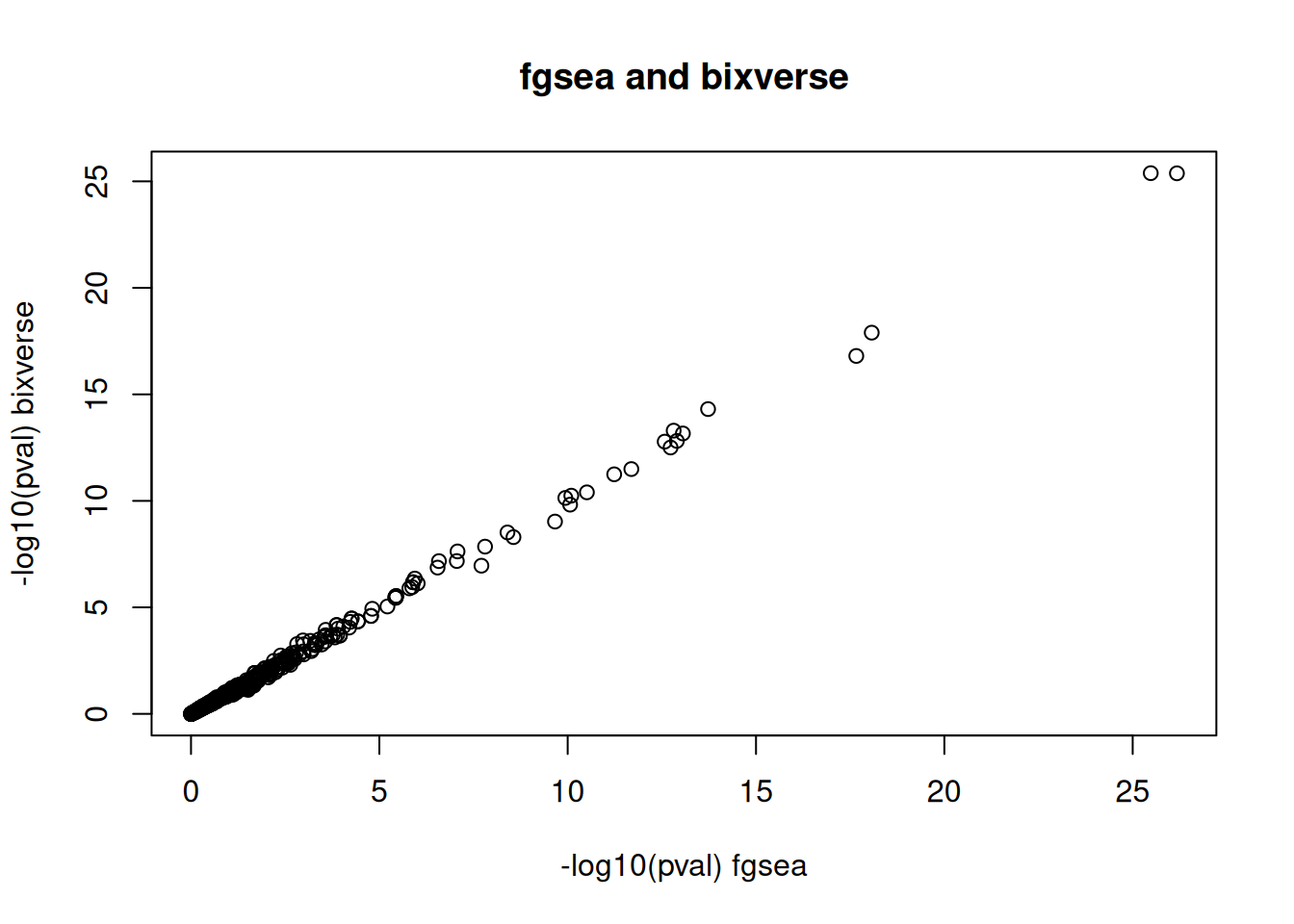
#> 6: 60406,20018,245688,20017 237 0.08175156The p-values from both implementations are in close agreement, as expected:
plot(
x = -log10(fgsea_res$pval),
y = -log10(bixverse_fgsea$pvals),
xlab = "-log10(pval) fgsea",
ylab = "-log10(pval) bixverse",
main = "fgsea and bixverse"
)
The Rust implementation is consistently faster than fgsea’s Rcpp backend — not by an enormous margin on small data (roughly 20% in typical benchmarks), but the gap widens with the number of pathways and permutations:
microbenchmark::microbenchmark(
fgsea = fgsea(
pathways = examplePathways,
stats = exampleRanks,
minSize = 15,
maxSize = 500
),
rust = calc_fgsea(
stats = exampleRanks,
pathways = examplePathways,
gsea_params = params_gsea(min_size = 15L)
),
times = 5L
)
#> Unit: seconds
#> expr min lq mean median uq max neval
#> fgsea 2.40426 2.438197 2.613924 2.576308 2.679570 2.971285 5
#> rust 2.16457 2.179880 2.195300 2.205283 2.208558 2.218208 5GO-aware GSEA: the elimination method
bixverse combines the elimination method with GSEA, using the same GeneOntologyElim object as the hypergeometric variant. The ontology is traversed leaf-first; for each term a permutation-based p-value is computed and if it falls below the elimination threshold, genes from that term are removed from all ancestors before they are tested. Once the full traversal is complete, the fgsea multilevel method is applied to terms that reached nominal significance to obtain more precise p-values than pure permutation alone can provide.
gene_universe_go <- unique(
unlist(go_data_dt[, "ensembl_id"], use.names = FALSE)
)
set.seed(42L)
random_stats <- rnorm(length(gene_universe_go))
names(random_stats) <- gene_universe_go
go_gsea_res <- fgsea_go_elim(
object = go_data_s7,
stats = random_stats
)
head(go_gsea_res)
#> go_id es nes size pvals n_more_extreme
#> <char> <num> <num> <num> <num> <num>
#> 1: GO:0031465 -0.9025468 -1.925965 6 6.165332e-05 0
#> 2: GO:0050877 0.5116421 1.950886 44 2.410338e-04 2
#> 3: GO:0072675 -0.8744037 -1.865910 6 2.625170e-04 0
#> 4: GO:0006066 -0.6718553 -2.026201 18 2.712234e-04 0
#> 5: GO:0071392 -0.5217830 -1.904151 36 3.788231e-04 0
#> 6: GO:0002090 -0.8302363 -1.868975 7 6.827804e-04 3
#> leading_edge
#> <list>
#> 1: ENSG00000110844,ENSG00000180370,ENSG00000160685
#> 2: ENSG00000131263,ENSG00000229674,ENSG00000172987,ENSG00000108878,ENSG00000050165,ENSG00000143061,...[19]
#> 3: ENSG00000170613
#> 4: ENSG00000287395,ENSG00000139354,ENSG00000211659,ENSG00000160345,ENSG00000007237,ENSG00000139679,...[7]
#> 5: ENSG00000118193,ENSG00000134531,ENSG00000166897,ENSG00000162980,ENSG00000170348,ENSG00000103018,...[17]
#> 6: ENSG00000061938,ENSG00000169507,ENSG00000289721,ENSG00000083814
#> log2err fdr go_name
#> <num> <num> <char>
#> 1: 0.5384341 0.4815124 Cul4B-RING E3 ubiquitin ligase complex
#> 2: 0.5188481 0.5295637 nervous system process
#> 3: 0.4984931 0.5295637 osteoclast fusion
#> 4: 0.4984931 0.5295637 alcohol metabolic process
#> 5: 0.4984931 0.5917217 cellular response to estradiol stimulus
#> 6: 0.4772708 0.8290480 regulation of receptor internalization